
Фармаконагляд — наука, на яку покладено завдання щодо виявлення, оцінки, розуміння та попередження несприятливих ефектів лікарських засобів або інших пов’язаних із цим проблем. Потреба в моніторингу безпеки виникла через нещасні випадки з летальним кінцем, спричинені анестезією, і вродженими вадами розвитку внаслідок використання талідоміду. На важливості фармаконагляду вперше було наголошено в 1848 р., коли дівчина на ім’я Ханна Грінєр з Англії померла після введення хлороформу для анестезії з метою видалення інфікованого нігтя на нозі. Через занепокоєння щодо безпеки використання анестетиків Lancet створив комісію для вирішення цієї проблеми, заохочуючи лікарів повідомляти про смерті, спричинені анестезією.

Г.А. Кордеро, MD, начальник відділу фармаконагляду, уповноважена особа, відповідальна за фармаконагляд, клінічний експерт АТ «Фармак», тренер «СтТР», www.sttd.com.ua https://www.linkedin.com/in/galyna-cordero-11a4287a/
Будь-яка фармакологічна речовина, котру вивчають як «кандидата у лікарські препарати», одночасно є і «реагентом» для дослідження функціонування біологічних систем у нормі та за різних патологій. На жаль, у деяких випадках такі «дослідження» призводять до трагічних наслідків у клінічній практиці, як, наприклад, «талідомідова трагедія». Необхідно, однак, підкреслити, що компанія «Хемі Грюненталь», яка вивела даний препарат на ринок наприкінці50-х років минулого століття, вивчала його безпеку відповідно до стандартів, що існували на той час.
Однак на мишах та щурах талідомід не мав тератогенної дії, яку було виявлено згодом, під час його широкого застосування в медичній практиці. Наслідком цього стала вимога щодо обов’язкового тестування тератогенності не менш ніж на трьох видах експериментальних тварин (зокрема, тератогенний ефект талідоміду спостерігали в експерименті на кролях).
У 1961 р. Макбрайд з Австралії написав до Lancet, повідомляючи про свою підозру щодо прийому талідоміду в період вагітності, що спричинило збільшення кількості вроджених вад розвитку у немовлят. Талідомід було представлено на ринку в 1957 р. для зменшення ранкової нудоти, виробник вважав його цілком безпечним для використання у вагітних. Однак застосування препарату призвело до виникнення аномалій розвитку плода та деформацій кінцівок (фокомелія) в 46 країнах світу, що засвідчило важливість моніторингу безпеки лікарських засобів після виходу на ринок.
«Талідомідова трагедія» спонукала до формування Міжнародної програми з моніторингу лікарських засобів Всесвітньої організації охорони здоров’я та до зміцнення нормативно-правової бази безпеки ліків. Після цього інциденту спонтанні повідомлення про побічні реакції на лікарські засоби (ПЛР) стали систематичними, організованими та регламентованими.
Прикладами у сфері фармаконагляду є проведення багатьох досліджень на підставі вивчення ефектів талідоміду, зокрема тератогенного і протипроказного, а також створення понад 30 окремих теорій про причини їхнього виникнення. З часом чимало таких досліджень сприяли розумінню механізмів, що лежать в основі ангіогенезу та розвитку кінцівок загалом, а також відкриттю антиканцерогенного ефекту талідоміду.
В сучасному фармаконагляді спостереження за ПЛР стали підґрунтям загального прогресу розуміння медицини окремо від безпеки фармакотерапії.
Під час перших хвиль пандемії відсутність вакцин і ліків для лікування/запобігання COVID-19 призвела до поспішної заміни показань вже схвалених ліків іншими показаннями. Внаслідок цього велику кількість ліків (наприклад, гідроксихлорохін, івермектин та азитроміцин) для лікування хворих наCOVID-19 почали використовувати не за призначенням, навіть якщо основні наукові докази їхніх переваг були низької якості на основі досліджень in vitro.
Моніторинг фармаконагляду в цьому контексті був вирішальним для визначення пов’язаних ризиків щодо препаратів, які використовуються не за призначенням, тим самим нагадуючи про принцип «спочатку не нашкодь», особливо якщо немає або є слабкі докази їхньої користі. Це стосується, зокрема, азитроміцину, макролідного антибіотика, який широко використовували для лікування пацієнтів із COVID-19. Його відома проаритмогенна активність, яка може посилюватися у разі застосування в комбінації з іншими препаратами, запропонованими для лікування COVID-19 (наприклад, гідроксихлорохін), стала підставою для регуляторних органів видавати застереження щодо його призначення, окрім випадків виникнення бактеріальної суперінфекції.
Індивідуальні звіти про ПЛР зберігаються в глобальній базі даних, і ними можна послуговуватися для встановлення причинно-наслідкових зв’язків між різними лікарськими засобами та пов’язаними з ними побічними реакціями. Клініцисти відіграють важливу роль у розпізнаванні та обов’язковому повідомленні про ПЛР представників регуляторних органів та заявників лікарських засобів.
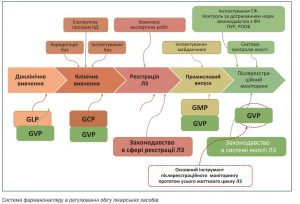
Фармаконагляд є необхідним протягом усього життєвого циклу лікарського засобу, включаючи всі фази, починаючи з доклінічних досліджень, клінічних досліджень, виробництва та дистриб’юції, щоб знизити ризики щодо їхньої безпеки.
Небезпечні фактори (ризики),що виникають у процесі життєвого циклу лікарських засобів:
- втрата якісних характеристик під впливом чинників навколишнього середовища;
- наявність у складі несумісних активних речовин;
- наявність у складі заборонених до застосування фарбуючих та допоміжних речовин;
- контамінація (забруднення) лікарських засобів;
- відсутність достовірної інформації про лікарський засіб.
Під час розробки препарату його безпеку досліджують на різних етапах. На доклінічному етапі основною метою оцінки безпеки є визначення безпечної дози для людей на підставі параметрів безпеки для клінічного моніторингу.
Вимоги щодо безпеки проведення доклінічних досліджень лікарських засобів
- Доклінічні (неклінічні) дослідження проводять з метою оцінки безпеки нової біологічно активної речовини або готового лікарського засобу, у складі якого вона міститься.
- Доклінічні (неклінічні) дослідження проводять на етапах розробки лікарських субстанцій та готової лікарської форми. На стадії доклінічних (неклінічних) досліджень комплекс заходів щодо безпеки включає систематизацію, аналіз та оцінку даних щодо:
- токсичності субстанції та готової лікарської форми після одноразового застосування;
- токсичності субстанції та готової лікарської форми після багаторазового застосування (підгострій та хронічній);
- репродуктивної токсичності (фертильності, ембріо- та фетотоксичності, тератогенності, пери- та постнатальної токсичності) субстанції та готової лікарської форми;
- генотоксичності субстанції та готової лікарської форми;
- мутагенного потенціалу субстанції та готової лікарської форми;
- канцерогенного потенціалу субстанції та готової лікарської форми;
- токсикокінетики субстанції та готової лікарської форми;
- фармакодинаміки субстанції та готової лікарської форми з метою виявлення ПЛР;
- місцевої переносимості субстанції та готової лікарської форми, зокрема фототоксичність, подразнювальну дію, місцеву гіперчутливість; можливості розвитку звикання та/або синдрому відміни ліків після застосування субстанції або готового лікарського препарату;
- ризику недостатньої фармакологічної ефективності субстанції та готового лікарського препарату;
- ризику реверсії у вихідну токсичну форму нетоксичного біологічного компонента;
- ризику фармацевтичної та фармакологічної несумісності з іншими лікарськими засобами.
На клінічній фазі дослідження І фази призначені для оцінки переносимості діапазону доз, які, як очікується, знадобляться для подальших клінічних досліджень у здорових добровольців. Дослідження II фази зосереджені на визначенні відповідного діапазону доз ліків у пацієнтів із захворюванням або станом, що представляє інтерес, тоді як клінічні випробування III фази є найважливішими дослідженнями, завдяки результатам яких потрібно вдосконалити розуміння профілю користі та ризику препарату та визначити менш поширені ПЛР. Хоча оцінка безпеки ліків є дуже суворою процедурою на етапі клінічних випробувань перед виходом на ринок, однак існують внутрішні обмеження, які не дозволяють якісно та ретельно оцінити профіль безпеки лікарського засобу.
Клінічні дослідження проводять за участі обмеженої кількості пацієнтів, вибраних із дотриманням суворих критеріїв прийнятності, тому вони не повністю представляють реальне населення та мають обмежену тривалість, що ускладнює виявлення рідкісних і тривалих ПЛР.
Вимоги до безпеки процесу проведення клінічних досліджень та (або) випробувань лікарських засобів
На стадії клінічних досліджень комплекс заходів щодо безпеки лікарського засобу передбачає оцінку:
- результатів хімічних, біологічних і фармацевтичних досліджень;
- результатів доклінічних (неклінічних) досліджень;
- дотримання протоколу клінічного дослідження;
- брошури дослідника та інших необхідних документів;
- звіту клінічного дослідження.
Від моменту створення препарату до його вірогідного виходу на ринок існують численні можливості для виникнення непередбачених проблем щодо безпеки. Це може бути пов’язане з хімічним складом препарату, його діючою речовиною, взаємодією з іншими ліками, помилками під час застосування в медичній практиці або наявністю специфічних особливостей пацієнта. Система фармаконагляду постійно відстежує ПЛР та надає відповідні відомості. Оперативне визначення потенційних ризиків дозволяє власникам регуляторних посвідчень, медичним працівникам і представникам регулюючих органів вжити необхідних заходів для зменшення шкоди пацієнтам.
Фармакологія безпеки — це оцінка та вивчення фармакологічних ефектів потенційного препарату (або допоміжної речовини), які не пов’язані з бажаним терапевтичним ефектом і тому можуть становити небезпеку, зокрема в осіб із порушенням або обмеженням однієї чи більше функцій організму. На відміну від інших доклінічних оцінок безпеки препарату, їх зазвичай проводять за умови застосування препарату в дозах, близьких до таких у разі призначення в клінічній практиці.
Завдання фармакологічних досліджень безпеки препарату:
- визначити небажані фармако-динамічні властивості;
- оцінити несприятливі фармако-динамічні та/або патофізіологічні ефекти;
- вивчити механізм наявних несприятливих фармакодинамічних ефектів та/або таких, що підозрюють.
Фармакологічне дослідження безпеки є дисципліною, яка постійно розвивається у фармацевтичній галузі. Неочікувані ефекти нових препаратів-кандидатів на функції основних органів (тобто вторинні фармакологічні ефекти) критично оцінюють на різноманітних моделях in vitro та на тваринах.
Фармаконагляд не обмежується лише моніторингом безпеки лікарських засобів — це також поширюється на збереження якості та стабільності фармацевтичних продуктів. На етапах виробництва та застосування зміни в процесі виробництва можуть вплинути на безпеку та ефективність препарату. Належні практики фармаконагляду допомагають підтримувати цілісність ліків, гарантуючи, що кожна подальша доза є такою самою безпечною та ефективною, як і попередня. Це зобов’язання щодо якості узгоджується з основними принципами догляду, орієнтованого на пацієнта.
Безпека лікарських засобів у процесі їхнього виробництва та виготовлення має ґрунтуватись на:
- виборі технологічних процесів та режимів їх здійснення на всіх етапах (дільницях) виробництва лікарських засобів;
- необхідній кваліфікації персоналу, який має достатній практичний досвід;
- виборі оптимальної послідовності технологічних процесів, що виключає забруднення та перехресне забруднення вироблених лікарських засобів;
- контролі роботи технологічного обладнання;
- належному утриманні виробничих приміщень, технологічного обладнання та інвентарю, які використовують в процесі виробництва лікарських засобів, у стані, що унеможливлює забруднення та перехресне забруднення лікарських засобів;
- виборі ефективних способів і періодичності санітарної обробки та очищення виробничих приміщень, технологічного обладнання та інвентарю, що використовують в процесі виробництва лікарських засобів. Планування приміщень та конструкція обладнання повинні мінімізувати ризик помилок, передбачати проведення ефективного прибирання та обслуговування з метою запобігання перехресному забрудненню та усунення будь-якого фактора, що погіршує якість продукції;
- здійсненні всіх необхідних видів контролю сировини, напівпродуктів і готових лікарських засобів відповідно до вимог технологічного процесу та встановленої нормативної документації;
- дотриманні умов зберігання сировини, фармацевтичних субстанцій і допоміжних речовин, що використовують для виробництва лікарських засобів;
- веденні та зберіганні документації, що підтверджує виконання вимог цього регламенту.
Вимоги до безпеки процесу зберігання та транспортування лікарських засобів
Умови зберігання лікарських засобів повинні гарантувати збереження властивостей, безпеку лікарських засобів протягом усього терміну їхньої придатності, запобігання контамінації, перехресній контамінації та пересортиці.
Умови збереження і транспортування лікарських засобів:
- контроль можливості їхньої ідентифікації та оцінки безпеки;
- відсутність контамінованості іншими лікарськими засобами (дозуваннями) і речовинами та впевненість у тому, що вони самі не контамінували;
- вживання відповідних запобіжних заходів для перешкоджання пошкодженням і розкраданням;
- захист та унеможливлення надмірного впливу чинників навколишнього середовища (температура, світло, воло-гість) та інших негативних факторів.
Збереження громадського здоров’я є першочерговою задачею системи охорони здоров’я, і фармаконагляд, зокрема, відіграє у цьому ключову роль. Систематично збираючи та аналізуючи дані про ПЛР та вивчаючи інші проблеми безпеки, фармаконагляд сприяє ширшому розумінню ризиків для здоров’я населення. Своєчасне втручання, наприклад відкликання продуктів або оновлення етикеток, вкладишів для пацієнтів та інструкцій для медичного застосування, може запобігти масштабній шкоді та зберегти довіру громадськості до системи охорони здоров’я та фармацевтичних продуктів.
Застосування фармаконагляду в клінічній практиці
У сфері охорони здоров’я фармаконагляд орієнтований на пацієнта. Ефективний фармаконагляд ґрунтується на послідовних високоякісних клінічних даних щодо ПЛР, особливо рідкісних побічних ефектів, які можуть потребувати міжнародних баз даних для виявлення проблем щодо безпеки. Однак наявні значні відмінності в міжнародних підходах до проведення фармаконагляду. Наприклад, у країнах Європи існує більша залежність від фінансування промисловості за постмаркетинговим наглядом, тоді як у Північній Америці це зазвичай відбувається через програми, що фінансує держава. У країнах Азії фармаконагляд менш розвинений і має значну варіабельність залежно від географічних, культурних та медичних особливостей кожного регіону.
Розпізнавання ПЛР та їхня диференціація з іншими станами або супутніми захворюваннями є складним завданням і вимагає від клініцистів знання клінічних і фармакологічних принципів ПЛР, зокрема про їхні типи, залежність від дози, реакції гіперчутливості, часові співвідношення та чинники ризику. Наприклад, віддалені ускладнення, такі як атипові переломи стегнової кістки, вторинні до використання бісфосфонатів, можуть виникнути лише за їхнього тривалого впливу. Підвищення ризику остеопорозу можливе, наприклад, після припинення прийому деносумабу.
Окрім усунення ускладнень, клініцисти також повинні консультувати пацієнтів для забезпечення постійного дотримання терапевтичного режиму і лікування основних станів та вести відповідну документацію щодо клінічних записів пацієнта, щоб уникнути подальшого впливу ліків. Клініцистів слід заохочувати повідомляти про ПЛР, щоб гарантувати, що профіль безпеки ліків реєструють на національному рівні, що допомагає у формулюванні регуляторних дій з метою мінімізації ризику для споживачів.
Більшість лікарів розуміють важливість і актуальність фармаконагляду в клінічній практиці. Однак викликає занепокоєння низька обізнаність щодо програм фармаконагляду та практичних аспектів у галузі безпеки лікарських засобів, зокрема куди і про що слід повідомляти з точки зору ПЛР. Виходячи з цього, нагальною потребою є впровадження необхідних освітніх та навчальних програм для поліпшення знань у галузі безпеки лікарських засобів, підвищення обізнаності та якості індивідуальних звітів щодо ПЛР. Цього можна досягти за допомогою навчання фармаконагляду невеликих інтерактивних навчальних груп у форматі лекцій і практичних демонстрацій у реальних ситуаціях з аналізом клінічних випадків.
Попереду ще багато роботи для створення навчальних програм та стандартизації компетенцій, необхідних для фармаконагляду. Функціональні та поведінкові компетенції мають поширюватися на різні рівні: клініцистам потрібно збирати інформацію або шукати докази про наявність ПЛР, потім їх обробляти та аналізувати, а уповноваженим особам— ухвалювати рішення щодо внесення будь-яких змін у результати на підставі виявлених ПЛР.
Набір основних компетенцій з метою проведення фармаконагляду включає такі навички для виявлення та запобігання побічним реакціям та іншим проблемам щодо безпеки лікарських засобів:
- аналітичні;
- оціночні;
- комунікативні;
- лідерства та системного мислення.
Продемонструвати ефективність фармаконагляду з точки зору впливу клініциста на безпеку пацієнтів досить складно. Наразі для цього використовують сурогатні маркери, такі як кількість і якість зареєстрованих ПЛР. Участь медичних працівників у фармаконагляді сприятиме позитивним змінам у практиках, пов’язаних із призначенням і спільним ухваленням терапевтичних рішень, а також у спілкуванні з пацієнтами. Проте необхідні подальші дослідження, щоб визначити та виміряти переваги додаткового залучення пацієнтів лікарями ланки первинної медичної допомоги з точки зору лікаря, який призначає лікарські засоби, та регуляторного органу.
Фармаконагляд передбачає забезпечення раннього виявлення нових ПЛР або формування підгруп пацієнтів із гіперчутливістю і пошук конкретних методів зниження таких ризиків. Зазвичай потрібні додаткові знання про ефективність та безпеку тривалого застосування препарату в комбінації з іншими лікарськими засобами в певних групах населення, таких як діти, вагітні жінки та пацієнти похилого віку.
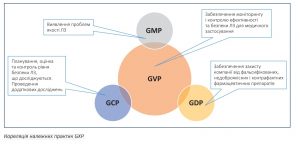
Мультидисциплінарність та трансдисциплинарність фармаконагляду
Фармаконагляд залишається, з одного боку, значною мірою ізольованою дисципліною, попри те, що як галузь дослідження вже включає численні дисциплінарні підходи, практики та сфери знань: клінічну практику, розробку лікарських засобів, доклінічні, клінічні, епідеміологічні, токсикологічні, фармакологічні дослідження, кризис-менеджмент, менеджерські навички. Для встановлення причинно-наслідкового зв’язку потрібно мати знання з диференціації проявів ПЛР із симптомами основного захворювання та можливими проявами ПЛР за медичного призначення інших лікарських засобів.
З іншого боку, трансдисциплінарність починається із загального сприйняття такої складної суспільної проблеми, як фармаконагляд. Трансдисциплінарні підходи можуть виходити за межі дисциплінарних методів і експертних знань, доповнюючи систему фармаконагляду. Через спільну мету це потребує колегіальної роботи представників незалежних дисциплін, щоб вийти за межі власних інтересів і вирішити проблему.
Трансдисциплінарність — це підхід, який дозволяє науці та іншим системам знань конструктивно взаємодіяти. Його сильна сторона полягає, по-перше, у тому, що із самого початку в нього залучені зацікавлені сторони, щоб допомогти вирішити нагальні питання, а по-друге — дозволяє уникнути пихи щодо того, які системи знань мають значення.
Трансдисциплінарність у галузі епідеміології дозволяє створювати кращі гіпотези і забезпечує засоби, за допомогою яких різнорідні методи та інтереси можуть бути зіставлені таким чином, що «ймовірно, стимулюватиме появу нових знань».
Трансдисциплінарність створює найкращу модель для розуміння природи проявів проблем безпеки лікарських засобів (ПЛР тощо) і спрямована на здійснення ефективного фармаконагляду. Висловлювання більш ефективних гіпотез, ширше залучення зацікавлених сторін із боку регуляторної спільноти, інтеграція різноманітних методів і джерел є важливими кроками до підвищення ефективності фармаконагляду. Фармаконагляд покликаний розглядати докази та вивчати можливість підвищення спроможності різноманітних груп поширювати сигнали про шкоду ПЛР. Отже, застосування міждисциплінарного підходу до фармаконагляду є довгостроковим завданням, яке потребує внесення структурних змін у медичну та фармацевтичну освіту, а також проведення доклінічних та клінічних досліджень на підприємствах фармацевтичної індустрії.
Поліпшення поширення знань та комунікації. Прозорість безпеки ліків
Прозорість має вирішальне значення для довіри до безпеки ліків. Проактивне та своєчасне інформування про нові (потенційні) ризики є важливим завданням, у вирішенні якого варто дотримуватись балансу між ранніми попередженнями та очікуванням переконливих доказів. Підвищення обізнаності без зайвого занепокоєння є ключовим аспектом комунікації ризиків, і не існує універсального рішення для всіх ризиків. Комунікаційний зміст стратегії має ґрунтуватися на достовірності сигналу, врахуванні наявних проблем, вивченні потенціалу для запобігання серйозним подіям, на застосуванні доступних альтернатив і врахуванні ризику небажаних ефектів.
Висновки
Фармаконагляд використовують для виявлення, оцінки, розуміння та запобігання ПЛР. Потреба в моніторингу безпеки виникла через нещасні випадки з летальним кінцем, спричинені анестезією, і вродженими вадами розвитку в наслідок використання талідоміду. Звіти про ПЛР зберігаються в глобальній базі даних і можуть бути використані для встановлення зв’язку між застосуванням різних ліків і пов’язаними з ними ПЛР. Клініцисти відіграють важливу роль у розпізнаванні та повідомленні про ПЛР до національних центрів фармаконагляду.
Попереду ще багато роботи для створення навчальних програм і стандартизації компетенцій, необхідних для фармаконагляду на принципах трансдисциплінарності та мультидисциплінарності. Функціональні та поведінкові компетенції мають ґрунтуватися на різних рівнях: клініцисти повинні збирати інформацію або докази щодо ПЛР, потім обробляти інформацію, а уповноважені особи — ухвалювати рішення щодо будь-яких змін у результатах, враховуючи ПЛР.
